



Medtec 2024暨第十八届国际医疗器械设计与制造技术展览会将于9月25-27日在上海世博展览馆开幕。9月25日0点前完成Medtec 2024预登记注册,可免费入场参观。

Medtec创新峰会2023开设医械创新产业链八大主题论坛,邀请来自中检院医疗器械检测所、国家药监局医疗器械审评中心、药监局医疗器械监管科学研究基地、中国科学院深圳先进技术研究院、全国医用电器标准化技术委员会、北京大学人民医院等50+......


专业轴承供应商SMB Bearings的总经理Chris Johnson解释了为什么正确选择滚珠和套圈材料以及合理的产品设计能够保证高精度轴承乃至医疗器械的长使用寿命。

导管鞘是经皮血管介入手术使用最多的通路器械,基本所有的经皮血管介入手术均需要通过导管鞘建立手术通路,其成功建立是手术成功的前提条件。因此,本文分享了关于导管鞘组制造指南的思考。



2024医疗器械展览会探秘“中国牙谷” 引领中国口腔产业新质生产力向新而发
据2024医疗器械展览会了解,在中国牙谷口腔专科医院建设项目、中国牙谷口腔产业园二期及配套基础设施建设项目火热建设现场,给人感受到中国牙谷高质量发展的脉动,映射出中国牙谷不断汇聚发展优势、增强发展动力的坚定决心。
Frost&Sullivan预计,2025年将达到157.26亿元,2021年-2025年复合年增长率为24.34%;2032年将达到419.73亿元,2025年-2032年复合年增长率为15.06%。
2024上海医疗器械创新展Medtec China探西门子医疗大动作!
2024年3月18日,西门子医疗(Siemens Healthineers)宣布,将关闭其基于PCR的快速诊断业务(Fast Track Diagnostics),同时公司预计裁员约90人(大部分员工在卢森堡工作),拟于2024年9月前完成。

全球骨科巨头公司Enovis公司,由并购之王丹纳赫的创始人两兄弟创办,有着传奇的发展历程(买来卖去)。2023年营业收入为17.07亿美元(约人民币123亿元),同比上涨9.22%。
2024医疗器械展会提醒:又一项新指导原则发布,助力哪些医疗器械细分领域崛起?
之前发布的《重磅!又一项新注册审查指导原则发布,创新医疗器械审评高质量发展再提速》和《速览|浅析医疗器械可用性工程的合规性》两篇文章中,我们介绍了可用性工程的基本概念、中外医疗器械可用性工程政策,并重点梳理了国家药监局器审中心3月19日发布的《医疗器械可用性工程注册审查指导原则》及其使用说明的内容要点。
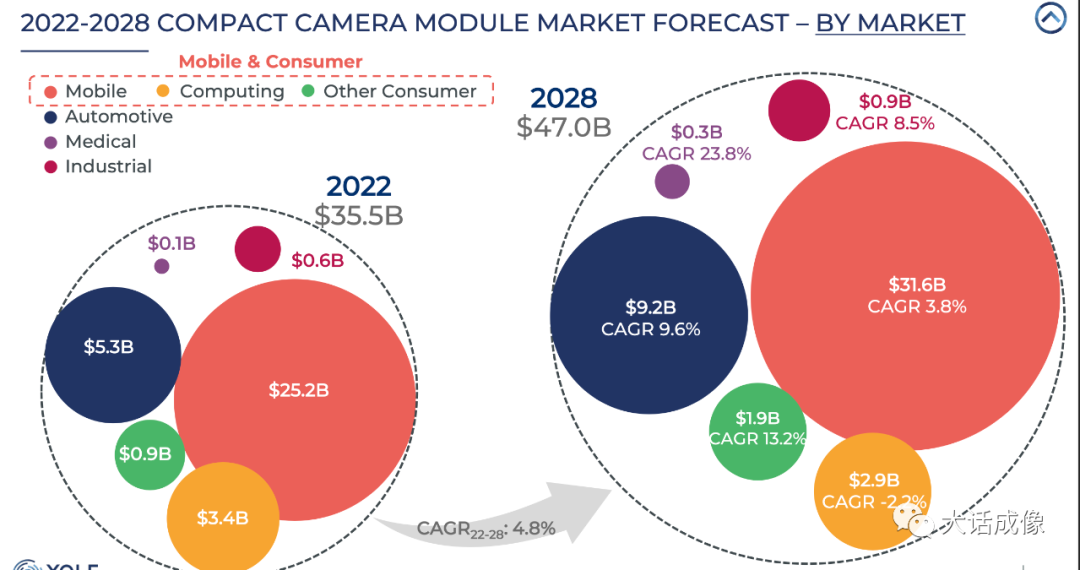
2024医疗器械展览会市场趋势调研之2023-2028年摄像头模组市场
本期的内容主要基于Yole发布的重磅报告《摄像头模块产业现状 2023》。接下来,我们将通过问答的形式,深入探讨摄像头模组市场的最新趋势和技术革新。

2024上海医疗器械展览会Medtec探秘开发中的新型人工晶体(一)
新型人工晶体有哪些在研方向呢?2024上海医疗器械展览会Medtec将会带你一探究竟。

21世纪以来,互联网、新能源、大数据等技术的迅猛发展,使得社会发生巨大的改变,生产工业发生重大变革。为应对全球挑战,我国根据发展的实际情况,提出《中国制造2025》的国家战略规划。毋庸置疑的是,智能制造必定成为世界制造业今后的发展趋势。当下目前国内智能制造的发展现状和未来的发展趋势如何呢?

2024上海医疗设备展心脏起搏器前沿:世界首款永动机式的无导线心脏起搏器
世界第一款通过能量收集装置达到能量自持的无导线起搏器,用“永动机”这个词是想表明该起搏器寿命很长(超过15年 vs 目前5-10年寿命),通过能量收集装置可以突破传统有导线或者最新无导线起搏器的寿命限制。
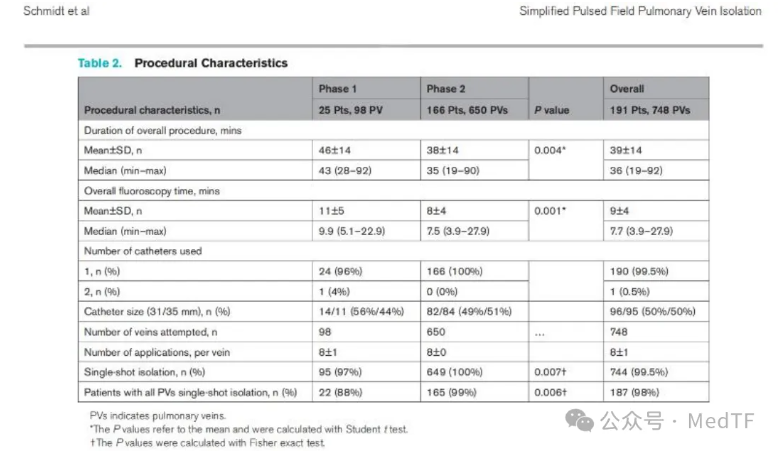
有限的内卷,无限的未来——医疗器械行业展览从导管设计再谈PFA
脉冲电场消融(PFA)是一种通过释放高压短程电脉冲能量,造成细胞膜不可逆电穿孔的非热消融技术,具有组织选择性,与传统的热消融原理(如射频、冷冻消融)有着本质区别。

上海医疗器械展会材料深度剖析|2024医用镍钛合金市场发展报告
镍钛合金是一种性能优异性能的高端合金,具有超强弹性等独特性能,在医疗器械等多个领域具有广泛应用。早在新冠流行之前,就一直是医疗器械的热门材料。
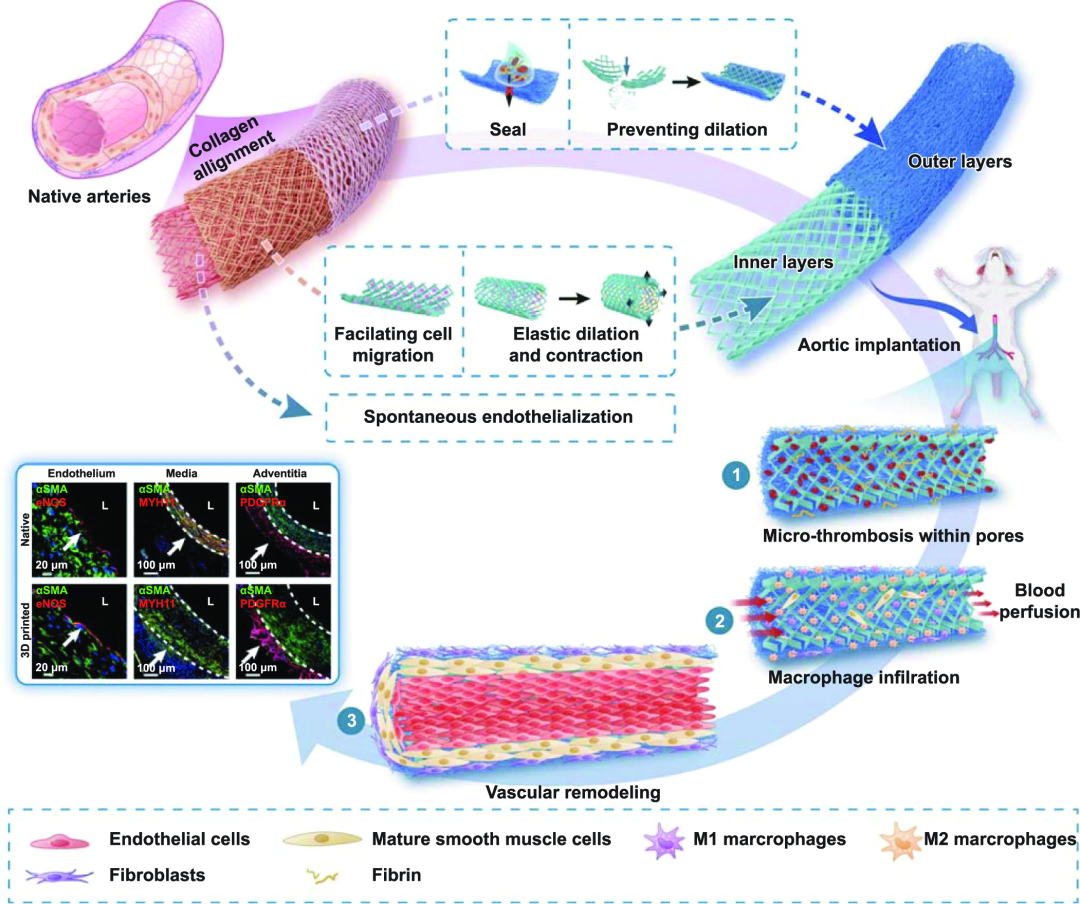
3D打印了高精度仿生的内外双层人工血管。该人工血管内层的有序结构,模拟了具有可收缩舒张的血管中膜。外层具有较高的拉伸模量,模拟了血管外膜,起到防止血管破裂、血液渗漏的作用。3D打印的人工血管植入体内1 个月后,在体内血流的刺激下,内层成功诱导了平滑肌层的快速重建及内皮化;而外层则诱导了血管外膜组织的再生,重塑了天然血管的三层结构,实现了血管在体内快速有序再生。

2024医疗器械展览会Medtec医用金属产品及服务合集,从原材料到高精密定制化生产加工(一)
9.25-27日,在上海世博展览馆1&2号馆举办的2024医疗器械展览会Medtec,将有古河科技材料 […]
Medtec思考究竟哪种注射笔设计可实现稳定可靠的给药精度?
本文详细介绍了,以给药剂量保持高度一致准确性为核心的注射笔设计,几个关键考虑因素,并分享了IDC在开发不同注射笔当中解决的一些主要问题。
在药品制造过程中,相当多的药品需要在洁净厂房内生产、包装,为保障生产过程中的环境符合规范,洁净厂房空调系统必须长期稳定运行。对空调系统的能耗改进,会带来显著的经济效益。目前,空调系统的能耗优化,主要从减少风机负荷、冷热交换器负荷来考虑。

2024医疗器械展览会智造聚焦:机器人关节如何做到精准控制的,内部复杂结构怎么设计?
现在工业机器人的自动化程度让人叹为观止,5轴6轴机器人具有如此多的关节,还能够做到运动和指令的精确传输,各部位紧密配合完成复杂的工作,让人不禁好奇它们的传动系统到底是怎样的,关节到底是什么结构的呢?
本文为学习性总结,主要源于审评五部赵艳红老师《含银盐敷料产品的技术审评关注点》培训,同时参考ISO/TR10993-22《医疗器械生物学评价第22部分纳米材料指南》、《含银敷料注册技术审查指导原则》(征求意见稿)》及国家食品药品监督管理总局关于规范含银盐医疗器械注册管理有关事宜的公告(2015年第225号),主要从含银敷料的监管要求、含银敷料技术评审关注点介绍。

本文重点从敷料类产品的简介、技术评审关注要点、常见共性问题和临床评价思考等方面介绍,以为诸君在研发过程中提供点滴参考。
医疗器械制造展Medtec解读:当一个器械中的“无声”变化导致与另一器械不兼容时
在设计一些医疗器械时,考虑系统安全工程的原则是一种很好的做法
医疗器械需要人机交互方能实现预期用途,这是医疗器械的显著特征之一。相关数据表明医疗器械使用问题较为突出,使用风险不容忽视,主要原因在于医疗器械可用性存在问题。
召回,收购 VS 集采,裁员:医疗器械制造展望2024医疗器械的何去何从
国产医疗器械,好像也走到了雁门关前。并无对错,不分新旧,都只是好奇:独上高楼,望尽天涯路。而路,在何方?
2024医疗器械展览会Medtec China提醒小巨人申报在即!三大利好助力企业蓬勃发展
参照2023年第五批专精特新“小巨人”的申报时间(2023年是3月15日至4月10日),预计第六批“小巨人”企业申报即将开启,申报企业需尽早确定自己的核心、主动产品的细分领域,做好审计规划,避免影响申报。
参与MedTech World全球系列医疗设计与技术展会,与2000+全球领先供应商面对面交流
合作媒体















参展商咨询:
Linc Cai 蔡锋
电话:+86 21 6157 7217
邮箱:[email protected]
研讨会咨询:
Rebecca Lv
电话:+86-21 6157 7279
邮箱: [email protected]
参观咨询:
Carina Li 李娜
电话:+86 10 6562 3308
邮箱: [email protected]
媒体联系:
Daisy Ding 丁小雨
电话:+86 10 6562 3307
邮箱:[email protected]
 X
X




